The BioChemical Library (BCL) is an open-source software package distributed under the permissive MIT license. Historically, it was developed to provide a broad array of unique tools for biological and chemical research, such as small molecule modeling, protein structure determination from sparse experimental data, machine learning, predictive modeling, and a broad array of cheminformatics tools.
With respect to biological modeling, the BCL contains the widely used secondary structure prediction program JUFO, a folding algorithm that assembles secondary structure elements, and loop construction tools that complete protein backbones. With this series of protocols the BCL allows construction of backbone models for large and membrane proteins from the primary sequence. Among its unique strengths are the incorporation of sparse and low resolution data into folding simulations.
With respect to cheminformatics, the BCL was written with with an emphasis on atom-level manipulation of molecules and strong support for graph comparisons, descriptor calculation, and quantitative structure-activity relationship modeling (QSAR). The BCL also contains a number of standard machine learning algorithms coded in C/C++ for the purposes of QSAR, including deep neural networks (DNN).
In recent years, the Rosetta software package has made substantial gains in protein structural modeling with and without experimental constraints. As of 2021, the deep learning algorithms AlphaFold2 and RoseTTAFold have fundamentally transformed protein structure prediction, making many of the early biology tools of the BCL obsolete. Moreover, the maturation of deep learning over the last decade in multiple areas of scientific investigation has resulted in an explosion of machine learning modalities available in general purpose software packages, such as TensorFlow and PyTorch. Thus, the addition of new machine learning algorithms into the BCL source code is no longer a priority as it was a decade ago. Instead, we are excited to embrace the open-source spirit of today's software communities as we work to integrate complementary software packges into the BCL, and vice versa.
Today, original code development in the BCL is primarily in the realm of small molecule drug design, cheminformatics, and enhanced user experience. This includes algorithms for reaction- and non-reaction-based design, molecule optimization routines, pharmacophore mapping, small molecule conformer generation, and interface code to increase interoperability with other software packages. We are also actively exploring creation of a graphical user interface (GUI) and accessibility of the BCL through a Python API. The most important thing to note, however, is the scope of the BCL is largely defined by the developers working on it, as well as ongoing changes to needs in the broader computer-aided drug discovery community.
The BCL is comprised of over one thousand classes organized in 42 namespaces with approximately 600,000 lines of code that form a collection of C++ algorithms developed since 2005. The original code that formed the foundation of the BCL was written in the 1990's and early 2000's by one of the original co-creators of the Rosetta macromolecular modeling software suite, Jens Meiler, and since then has been an academic project in the Meiler Lab at Vanderbilt University for graduate students, post-doctoral fellows, and scientific programmers. We are excited to take the next step and make the BCL openly available. It is our hope that this will both expand the user-base and contributor-based, as well as inject new ideas into the community.
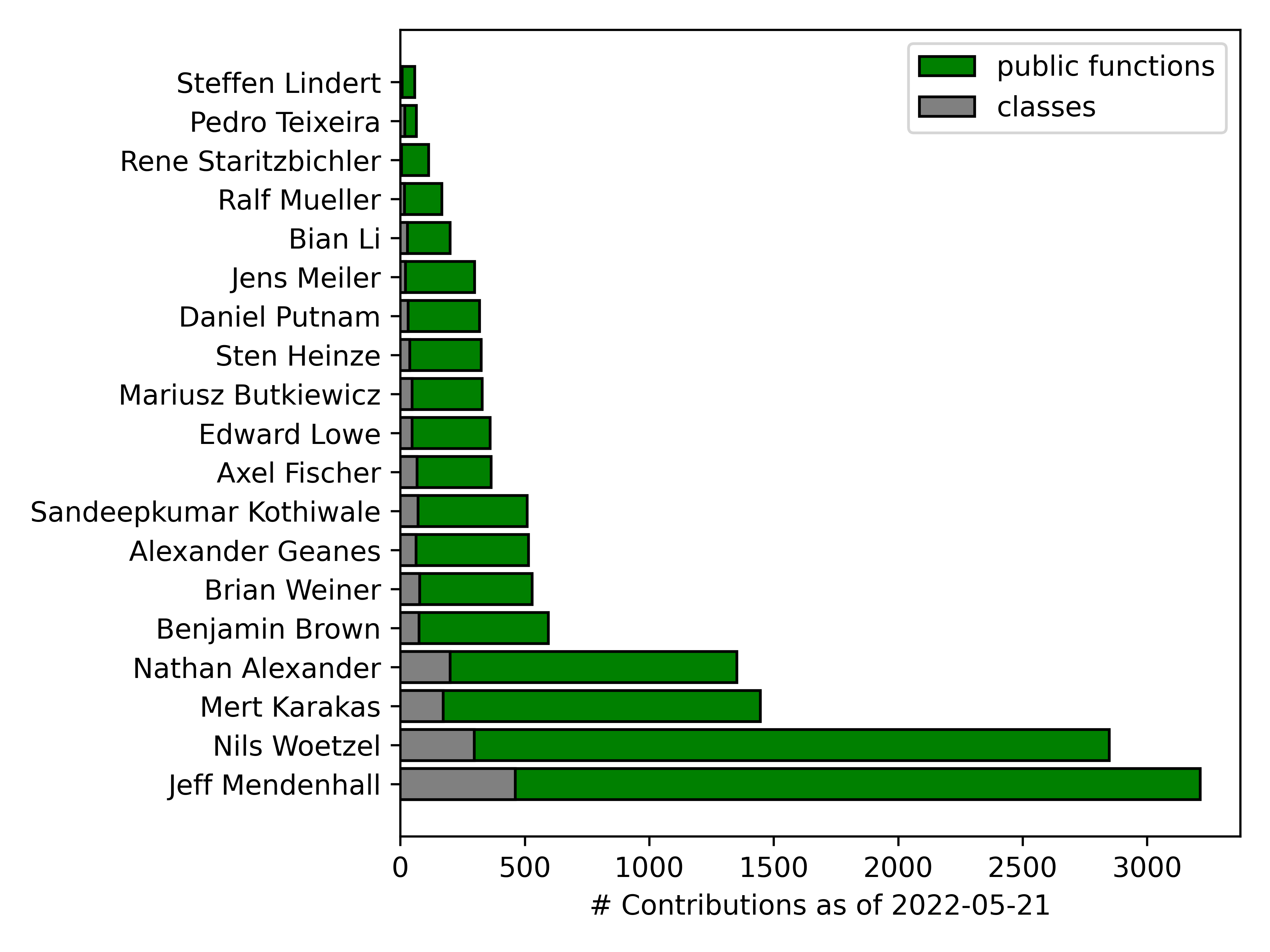
Trainees in the Meiler Lab have contributed to BCL development since the mid-2000's. It is difficult to measure the impact of individual developers on a large shared software project, especially as each cohort encountered unique challenges in solving their respective problems. Moreover, there have been dozens of smaller contributions over the years from high school students, undergraduate research assistants, rotation students, and of course graduate students, post-docs, programmers, and research faculty. Nonetheless, we would like to highlight our core development team. See below for tallied contributions from those who have written more than 50 public functions and more than 1 class in the BCL.
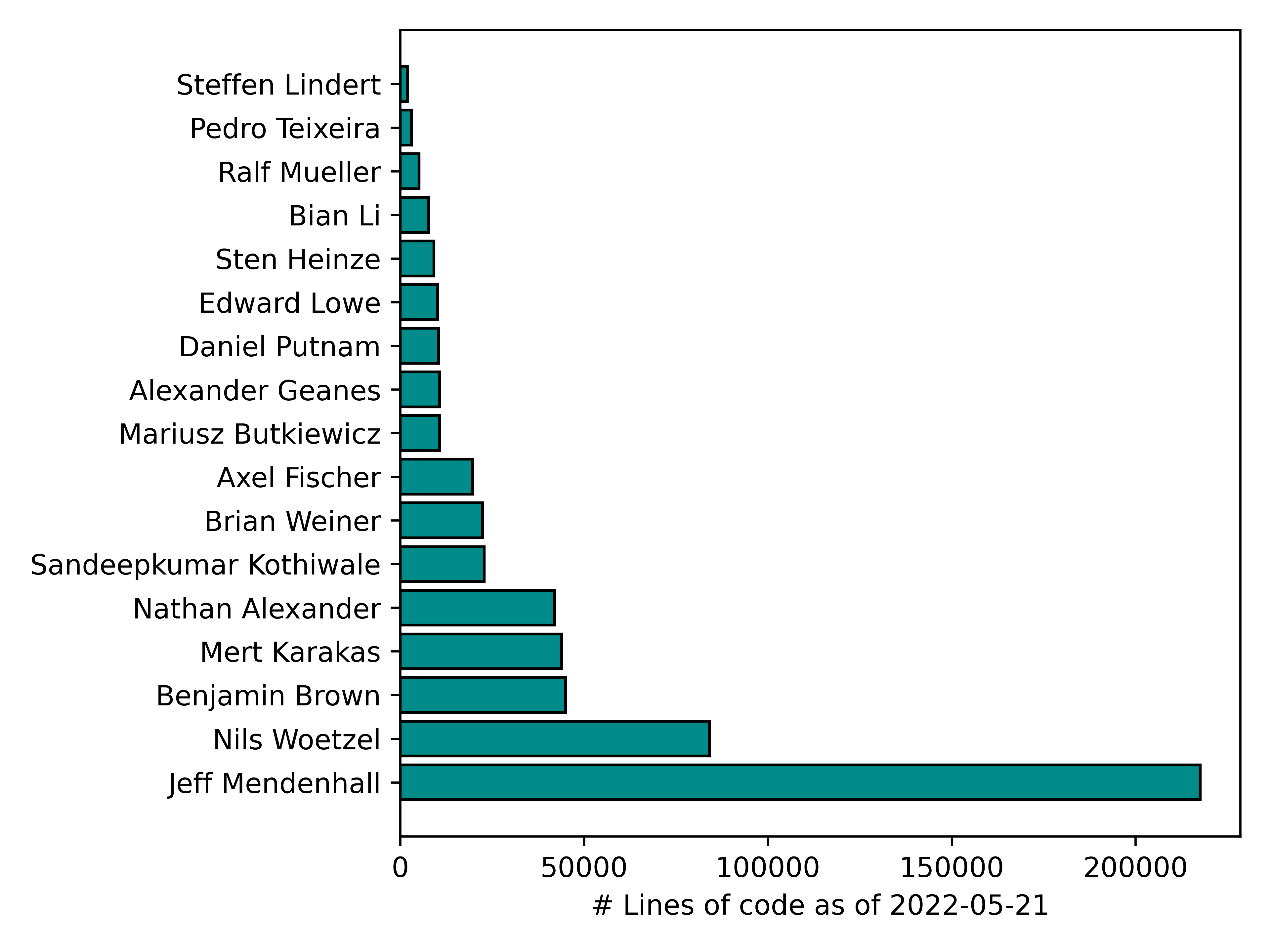
We can also plot developer contributions in lines of code (excluding comments).
If you plan to compile the BCL source code instead of using any of the available binaries, read COMPILING.md for detailed instructions.
If you wish to contribute to the BCL code base, please read through CODE_OF_CONDUCT.md and CONTRIBUTING.md beforehand. Discussions about new features, bug fixes and documentaion are held in the 'Issues' tab under the BCL GitHub (https://github.com/BCLCommons/bcl). All other discussions are held in the 'Discussions' tab on the BCL GitHub.