
With the rapidly increasing availability of High-Throughput Screening (HTS) data in the public domain, such as the PubChem database, methods for ligand-based computer-aided drug discovery (LB-CADD) have the potential to accelerate and reduce the cost of probe development and drug discovery efforts in academia. We assemble nine data sets from realistic HTS campaigns representing major families of drug target proteins for benchmarking LB-CADD methods. Each data set is public domain through PubChem and carefully collated through confirmation screens validating active compounds. These data sets provide the foundation for benchmarking a new cheminformatics framework BCL::ChemInfo, which is freely available for non-commercial use. Quantitative structure activity relationship (QSAR) models are built using Artificial Neural Networks (ANNs), Support Vector Machines (SVMs), Decision Trees (DTs), and Kohonen networks (KNs). Problem-specific descriptor optimization protocols are assessed including Sequential Feature Forward Selection (SFFS) and various information content measures. Measures of predictive power and confidence are evaluated through cross-validation, and a consensus prediction scheme is tested that combines orthogonal machine learning algorithms into a single predictor. Enrichments ranging from 15 to 101 for a TPR cutoff of 25% are observed.
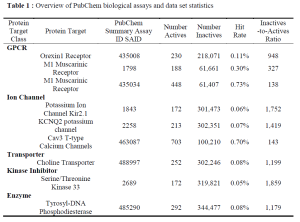
The active compounds of each data set utilized in this benchmark can be downloaded here:
The SD Files for inactive compounds of each data set are large in size. Please see the protocol capture to get the data.
Protocol Capture